
Page 118 - AN-4-1
P. 118
Advanced Neurology ADGRV1 and MED13L mutation
which can back normal. Whole-exome sequencing was 2) A heterozygous 7 base pair deletion in Exon 10 of
done, which revealed likely pathogenic mutations in two the MED13L gene in chromosome 12 (chr12:g.116
genes. 009074_116009080del; Depth: 141x) that results in
She underwent targeted gene sequencing. DNA a frameshift and premature truncation of 38 amino
extracted from blood was used to perform targeted gene acids downstream to codon 445 (p.Ser445GlnfsTer38;
capture using a custom capture kit. Clinically relevant ENST00000281928.9).
mutations in both coding and non-coding regions were Both of these variants have not been reported in the
annotated using published variants in the literature and 1000 genomes, gnomAD v3.1, gnomdAD v2, TOPMed,
the following set of disease databases: ClinVar, OMIM, and our internal databases. Subsequently, the proband
HGMD, LOVD, DECIPHER (population CNV), and sample along with samples from her parents were taken for
SwissVar. Two likely pathogenic variants were detected sanger validation and segregation analysis, which revealed
(Table 1). that the variant in the ADGRV1 gene was found to be
1) A heterozygous nonsense variant (c.1169dup) segregating in the heterozygous state in her unaffected
in exon 7 of the ADGRV1 gene in chromosome mother and that the variant in the MED13L gene was
5 (e (chr5:g.90627707dup; Depth: 24x) that results found to be de novo (Table 2).
in a stop codon and premature truncation at codon
390 (p.Tyr390Ter; ENST00000405460.9) 3. Discussion
MED13L-related intellectual disability is a rare syndrome
caused by loss-of-function intragenic variants or whole-
gene deletions in MED13L, with several missense variants
also reported recently. Although MED13L had initially
been believed to be a single gene cause of complex cyanotic
heart disease, it had later been shown to have a broader
8,9
clinical spectrum involving intellectual disability, speech
impairment, and behavioral issues. The most common
2
dysmorphic features reported are a bulbous nasal tip,
open mouth appearance, low set ears, and a broad nasal
bridge. This characteristic facial dysmorphism along with
2
speech impairment was noted in our patient. A review of
the literature found that very few of these cases developed
refractory epilepsy, all of whom had corresponding missense
mutations. In a study of 37 patients, Smol et al. found that
3
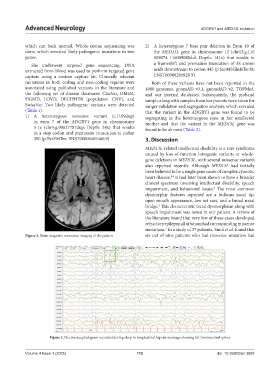
Figure 2. Brain magnetic resonance imaging of the patient six out of nine patients who had missense mutation had
Figure 3. Electroencephalogram recorded during sleep in longitudinal bipolar montage showing left frontocentral spikes
Volume 4 Issue 1 (2025) 112 doi: 10.36922/an.3602