
Page 95 - AN-3-2
P. 95
Advanced Neurology Genetics of neurodevelopmental disorders in Mexico
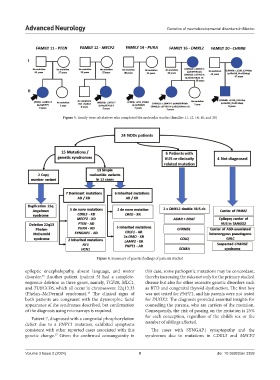
Figure 3. Family trees of relatives who completed the molecular studies (families 11, 12, 14, 16, and 20)
Figure 4. Summary of genetic findings of patients studied
epileptic encephalopathy, absent language, and motor this case, some pathogenic mutations may be concordant,
disorder. Another patient (patient 5) had a complete- thereby increasing the risks not only for the primary studied
15
sequence deletion in three genes, namely, TCF20, MLC1, disease but also for other recessive genetic disorders such
and TUBGCP6, which all occur in chromosome 22q13.33 as BTD and congenital thyroid dysfunction. The first boy
16
(Phelan–McDermid syndrome). The clinical signs of was not tested for PNPT1, and his parents were not tested
both patients are congruent with the dysmorphic facial for DUOX2. The diagnosis provided essential insights for
appearance of the syndromes described, but confirmation counseling the parents, who are carriers of the mutation.
of the diagnosis using microarrays is required. Consequently, the risk of passing on the mutation is 25%
for each conception, regardless of the child’s sex or the
Patient 7, diagnosed with a congenital phosphorylation
defect due to a PNPT1 mutation, exhibited symptoms number of siblings affected.
consistent with other reported cases associated with this The cases with SYNGAP1 synaptopathy and the
genetic change. Given the confirmed consanguinity in syndromes due to mutations in CDKL5 and MECP2
17
Volume 3 Issue 2 (2024) 9 doi: 10.36922/an.3359