
Page 68 - GPD-1-1
P. 68
Gene & Protein in Disease A novel USH2A gene mutation in retinitis pigmentosa
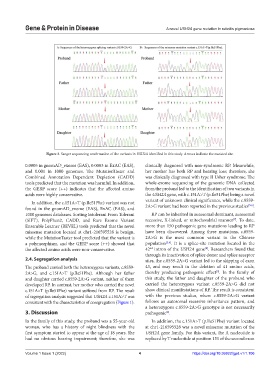
Figure 3. Sanger sequencing confirmation of the variants in USH2A identified in this study. Arrows indicate the mutated site.
0.0005 in gnomAD_exome (EAS), 0.0003 in ExAC (EAS), clinically diagnosed with non-syndromic RP. Meanwhile,
and 0.001 in 1000 genomes. The MutationTaster and her mother has both RP and hearing loss; therefore, she
Combined Annotation Dependent Depletion (CADD) was clinically diagnosed with type II Usher syndrome. The
tools predicted that the mutation was harmful. In addition, whole-exome sequencing of the genomic DNA collected
the GERP score (++) indicates that the affected amino from the proband led to the identification of two variants in
acids were highly conservative. the USH2A gene, with c.151A>T (p.Ile51Phe) being a novel
In addition, the c.151A>T (p.Ile51Phe) variant was not variant of unknown clinical significance, while the c.8559-
[5,6]
found in the gnomAD_exome (EAS), ExAC (EAS), and 2A>G variant had been reported in the previous studies .
1000 genomes databases. Sorting Intolerant From Tolerant RP can be inherited in autosomal dominant, autosomal
(SIFT), PolyPhen2, CADD, and Rare Exome Variant recessive, X-linked, or mitochondrial manner . To date,
[7]
Ensemble Learner (REVEL) tools predicted that the novel more than 150 pathogenic gene mutations leading to RP
missense mutation located at chr1-216595528 is benign, have been discovered. Among these mutations, c.8559-
while the MutationTaster tool predicted that the variant is 2A>G is the most common variant in the Chinese
a polymorphism, and the GERP score (++) showed that population [5,6] . It is a splice-site mutation located in the
[8]
nd
the affected amino acids were non-conservative. 42 intron of the USH2A gene . Researchers found that
through its inactivation of splice donor and splice acceptor
2.4. Segregation analysis sites, the c.8559-2A>G variant led to the skipping of exon
The proband carried both the heterozygous variants, c.8559- 43, and may result in the deletion of 41 amino acids,
[9]
2A>G, and c.151A>T (p.Ile51Phe). Although her father thereby producing pathogenic effect . In the family of
and daughter carried c.8559-2A>G variant, neither of them this study, the father and daughter of the proband who
developed RP. In contrast, her mother who carried the novel carried the heterozygous variant c.8559-2A>G did not
c.151A>T (p.Ile51Phe) variant suffered from RP. The result show clinical manifestations of RP. The result is consistent
of segregation analysis suggested that USH2A c.151A>T was with the previous studies, where c.8559-2A>G variant
consistent with the characteristics of cosegregation (Figure 1). follows an autosomal recessive inheritance pattern, and
a heterozygous c.8559-2A>G genotype is not necessarily
3. Discussion pathogenic .
[8]
In the family of this study, the proband was a 55-year-old In addition, the c.151A>T (p.Ile51Phe) variant located
woman, who has a history of night blindness with the at chr1-216595528 was a novel missense mutation of the
first symptom started to appear at the age of 18 years. She USH2A gene family. For this variant, the A nucleotide is
had no obvious hearing impairment; therefore, she was replaced by T nucleotide at position 151 of the second exon
Volume 1 Issue 1 (2022) 4 https://doi.org/10.36922/gpd.v1i1.106