
Page 121 - ITPS-7-1
P. 121
INNOSC Theranostics and
Pharmacological Sciences Repurposed Drugs as inhibitors of Pfmrk
A B
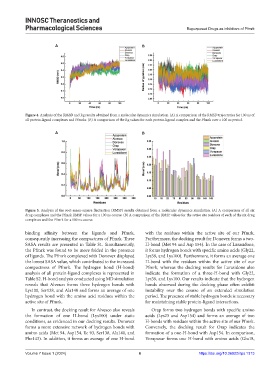
Figure 4. Analysis of the RMSD and Rg results obtained from a molecular dynamics simulation. (A) A comparison of the RMSD trajectories for 100 ns of
all protein-ligand complexes and Pfmrks. (B) A comparison of the Rg values for each protein-ligand complex and the Pfmrk over a 100 ns period.
A B
Figure 5. Analysis of the root-mean-square fluctuation (RMSF) results obtained from a molecular dynamics simulation. (A) A comparison of all six
drug complexes and the Pfmrk RMSF values for a 100 ns course. (B) A comparison of the RMSF values for the active site residues of each of the six drug
complexes and the Pfmrk for a 100 ns course.
binding affinity between the ligands and Pfmrk, with the residues within the active site of our Pfmrk.
consequently increasing the compactness of Pfmrk. These Furthermore, the docking result for Donovex forms a two-
SASA results are presented in Table S1. Simultaneously, H-bond (Met 94 and Asp 154). In the case of Lurasidone,
the Pfmrk was found to be more folded in the presence it forms hydrogen bonds with specific amino acids (Gly22,
of ligands. The Pfmrk complexed with Donovex displayed Lys39, and Lys100). Furthermore, it forms an average one
the lowest SASA value, which contributed to the increased H-bond with the residues within the active site of our
compactness of Pfmrk. The hydrogen bond (H-bond) Pfmrk, whereas the docking results for Lurasidone also
analysis of all protein-ligand complexes is represented in indicate the formation of a three-H-bond with Gly22,
Table S2. H-bond analysis conducted using MD simulation Lys39, and Lys100. Our results indicate that the hydrogen
reveals that Alvesco forms three hydrogen bonds with bonds observed during the docking phase often exhibit
Lys100, Ser138, and Ala140 and forms an average of one instability over the course of an extended simulation
hydrogen bond with the amino acid residues within the period. The presence of stable hydrogen bonds is necessary
active site of Pfmrk. for maintaining stable protein-ligand interactions.
In contrast, the docking result for Alvesco also reveals Orap forms two hydrogen bonds with specific amino
the formation of one H-bond (Lys100) under static acids (Lys23 and Asp154) and forms an average of two
conditions, as evidenced in our docking results. Donovex H-bonds with residues within the active site of our Pfmrk.
forms a more extensive network of hydrogen bonds with Conversely, the docking result for Orap indicates the
amino acids (Met 94, Asp154, Ile 93, Ser138, Ala140, and formation of a one-H-bond with Asp154. In comparison,
Phe143). In addition, it forms an average of one H-bond Vorapaxar forms one H-bond with amino acids (Glu18,
Volume 7 Issue 1 (2024) 6 https://doi.org/10.36922/itps.1313